About DynDom3D
Back to DynDom3DBasic Method
DynDom3D is a new program for the analysis of domain movements in large, multi-chain, biomolecular complexes. The program is applicable to any molecule for which two atomic structures are available that represent a conformational change indicating a possible domain movement. Unlike the original DynDom (DynDom1D) the method is blind to atomic bonding and atom type and can therefore be applied to biomolecular complexes containing different constituent molecules such as protein, RNA or DNA. At the heart of the method is the use of blocks located at grid points spanning the whole molecule. The rotation vector for the rotation of atoms from each block between the two conformations is calculated. Treating components of these vectors as coordinates means that each block is associated with a point in a “rotation space” and that blocks with atoms that rotate together, perhaps as part of the same rigid domain, will have co-located points. Thus a domain can be identified from the clustering of points from blocks within it. Domain pairs are accepted for analysis of their relative movements in terms of screw axes based upon a set of reasonable criteria. The results provide a depiction of the conformational change within each molecule that is easily understood, giving a perspective that is expected to lead to new insights. It has basically five parameters: a minimum domain size (in number of atoms), a ratio of interdomain displacement to intradomain displacement, a grid length, a block factor, and a block occupancy percentage. Details of the program are described in the following paper:
DynDom3D Webserver
Pairs of multi-chained structures that may have a domain movement will often not have a one-to-one correspondence between the atoms as they are ordered in their respective PDB files. This webserver has a preprocessor which creates this one-to-one correspondence. This is complicated as these differences may occur at the model level, the chain level, the residue level or the atom level. The image below shows how models and chains are treated. Only chains within the same model are compared and it is presumed that corresponding chains (identified by chain terminators "TER") are ordered equivalently in each model. Residues within each chain are compared using sequence alignment and inserted residues in either chain are excised. Atoms within each aligned residue are aligned and inserted atoms in either residues are excised.
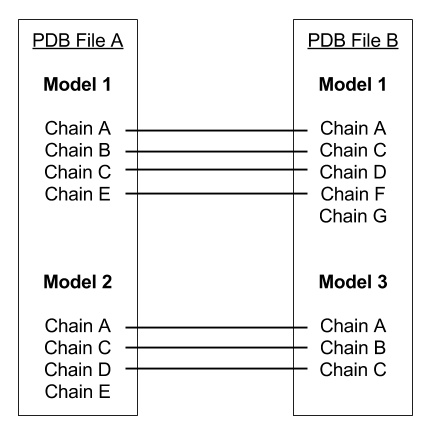 In this example, the residues under Chain E in Model 2 of PDB File A and, Chain G in Model 1 of PDB File B would not be present in the final PDB file.
In this example, the residues under Chain E in Model 2 of PDB File A and, Chain G in Model 1 of PDB File B would not be present in the final PDB file.
Input Parameters
At input you can specify four control parameters: the "Grid Size", g, the "Block Factor", ,b, the "Occupancy",occ, and the "Minimum Domain Size". A cubic grid of length g Angstroms spans one of the molecules and at each grid point there is a cubic block of length b×g where b is an integer. Movements of atoms within a block between the two structures are analysed to determine dynamic domains via the evaluation of rotation vectors. Both g and b determine the size of the blocks through b×g, but b determines the maximum number of overlapping grid cells which is given by b3-b2. The greater the number of overlapping grid cells, the greater the smoothing. Blocks on the surface may be sparsely occupied contributing noise. The quantity occ has a value between 0 and 1 and all blocks for which the number of atoms within the block is lower than occ×NMax are excluded from the analysis to determine dynamic domains using rotation vectors. The Minimum Domain Size is specified in number of atoms.
It has been found that results can be robust against variation of these parameters but if you get a null result (see below) using default values then you can try changing them.
Example Run

The top panel shows the input parameters used. The second panel is a JSmol graphics window containing the structure of conformer 1 with domain colouring. The "arrow molecules" depict the interdomain screw axes (hinge axes). The colour of the arrow shaft is the colour of the fixed domain (blue in this case) and the colour of the moving domain is given by the colour of the arrow head. This allows one to associate the pair of domains with the arrow that depicts their relative movement. The movement is a rotation about and a translation along the axis of the moving domain relative to the fixed domain with the direction of rotation given by the right-hand rule. The bottom panel gives details on the domains and the movement, including numbers of atoms the domains comprise, angles of rotations, etc.
Null Results
It is possible that you will get a null result. You can try altering the parameters slightly but persistent failure to get any result probably means that according to the criteria used by DynDom3D your structures do not have a domain movement. However, there are some issues with the DynDom3D program that we would like to resolve in time to improve it.