
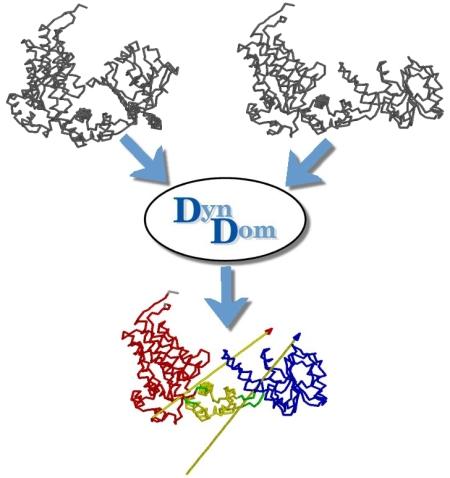
DynDom is a program to determine domains, hinge axes and hinge bending residues in proteins where two conformations are available.
This website provides access to the DynDom program and its associated databases. There are five databases: the user-created database that accumulates successful DynDom runs submitted by users, the non-redundant database that arose from an exhaustive study of X-ray structures in the PDB, a database of ligand-induced domain movements in enzymes, a database that classifies domain movements based on Dynamic Contact Graphs, and a database that classifies movements based on the effect on the domain interface.
DynDom6D is now available from the Download page.
The program analyses a pair of structures of a multimeric biomolecule to identify domain movements, providing a report on the relative domain movements and a PyMol session which shows the structures, domains and interdomain screw axes. This example output shows the domain movement between the open and closed states of the SARS-CoV-2 (coronavirus) spike glycoprotein.
For more information, see 'Methodological improvements for the analysis of domain movements in large biomolecular complexes'.
| Contents | |
|---|---|
| Dynamic Domain Analysis | |
| Program Description | |
| Databases | |
| References |

When to use DynDom
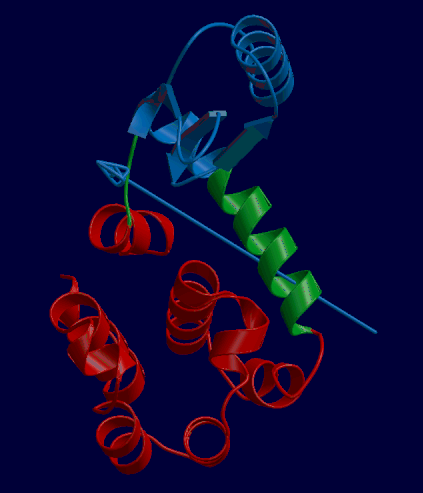
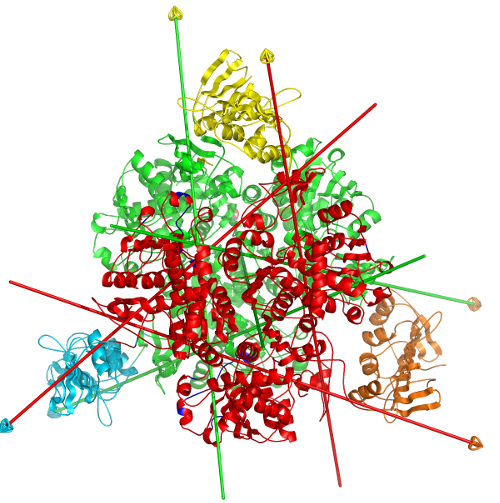
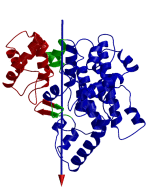
DynDom is a program that determines protein domains, hinge axes and amino acid residues involved in the hinge bending. It is fully automated. You can use DynDom if you have two conformations of the same protein. These may be two X-ray structures, or structures generated using simulation techniques such as molecular dynamics or normal mode analysis. The application of DynDom provides a view of the conformational change that is easily understood. The conformational change may be quite complicated in detail, but by using DynDom you can visualize it as involving the movement of domains as quasi-rigid bodies. The analysis of a conformational change in terms of domain movements only makes sense if the interdomain deformation is at least comparable to the intradomain deformation. You can use DynDom to assess this, but the results could be misleading if this is not the case. DynDom allows you to visualize the domain motion in terms of the rotation of one domain relative to another. Here we see the motion of the red domain relative to the blue domain that occurs upon substrate binding in citrate synthase. The arrow indicates the hinge axis and green those regions where the hinge bending takes place.
Determination of Dynamic Domains
The program initially determines the "dynamic domains." First, a whole protein best fit of the two conformations is made. Then, rotation vectors of short main-chain segments are determined. A clustering algorithm is then used to identify clusters of rotation vectors. Residues from segments in an individual cluster form a possible dynamic domain.

Determination of Hinge Axes
Groups of residues are only accepted for the analysis of hinge axes if they satisfy a criterion based on the ratio of the interdomain displacement to intradomain displacement with another group of residues with which there exists a physical connection. If this is the case the two groups of residues form dynamic domains and their interdomain motion is meaningful. The axes determined are in fact interdomain screw axes. This is based on the theorem of Chasles which states that the general displacement of a rigid body is a screw motion. The location of the interdomain screw axis tells us something about the kind of motion allowed by the interdomain connections. It is possible for the interdomain screw axis to be located far away from the interdomain connections if they are very flexible. Only if the interdomain screw axis is located near to those residues involved in the interdomain bending (defined below) can we think of the axis as a hinge axis. In such a case we call the axis an, "effective hinge axis" and the residues are said to be acting as "mechanical hinges."
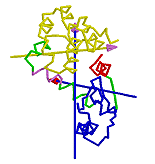
Determination of Residues Involved in Interdomain Bending
If one domain is fixed in space with the other rotating, then one will see a rotational transition in the connecting region between the two domains. One can define the residues involved in the interdomain bending to be those at the interdomain boundaries, as found by the clustering algorithm, plus those neighbouring residues whose rotations are outside the main distribution. If the clusters are treated as 3-dimensional normal distributions then bending residues are those that lie outside the ellipsoids of constant probability density that correspond to a significance value P of 0.2.
Closure and Twist Motion
Axes can be classified into two extreme types: those parallel to the line joining the centres of mass of a pair of domains, and those perpendicular to this line. The former are called twist axes, the latter, closure axes. Any axis can be decomposed into components parallel or perpendicular to this line and a percentage measure of the degree of closure motion can be defined from the square of the projection on the closure axis.
How DynDom Works
DynDom uses the K-means clustering algorithm to find clusters of rotation vectors. A cluster in rotation space may not correspond to a cluster in real space, but rather a fragmented region. Such a fragmented region one would not normally call a domain. DynDom splits up any clusters that do not correspond to heavy atoms connected through a network of distances of 4.0 angstrom or less, into domains. In order for DynDom to analyse domain pairs in terms of their interdomain movement two criteria must be satisfied. The first concerns the minimum domain size. If a domain comprises fewer residues than the minimum domain size set by the user, then segments from this domain are united with the larger domains they are embedded in. If all the domains from any single cluster are smaller than the minimum domain size, the program stops, unless this is the first cluster found (K=2). For every domain larger than the minimum size, the program checks which are connected directly through the backbone (not through another domain), and calculates the ratio of interdomain displacement to intradomain displacement for every connected pair. If this ratio is less than the user specified minimum (the second criterion) then this pair are not analysed. The program finds the largest number of clusters for which all connected domain pairs that satisfy both criteria. It is these domain pairs that are analysed in terms of interdomain screw axes, etc. If this is not possible it will analyse any domain pair for interdomain screw axes, etc, provided that the two criteria are satisfied
User-Created Database
This website allows users to select pairs of PDB files that may reveal a domain movement for input to the DynDom program. If the run is successful then the results are automatically loaded into the database along with all other successful runs. You can browse all the results and make simple searches. It allows us to tap the knowledge of individual specialists around the world to help populate a single database, which can act as a repository of information on protein domain movements.
See the following paper for details:
R.A.Lee, M.Razaz, S.Hayward,
"The DynDom database of protein domain motions"
Bioinformatics, 19, 10, 2003
ABSTRACT
The DynDom database of protein domain motions
Lee RA, Razaz M, Hayward S.
A relational database has been developed based on the results from the application of the DynDom program to a number of proteins for which multiple X-ray conformers are available. The database is populated via a web-based tool that allows visitors to the website to run the DynDom program server-side by selecting pairs of X-ray conformers by Protein Data Bank code and chain identifier.
Non-Redundant Database
An exhaustive analysis was carried out on all available protein structures in order to build a non-redundant and comprehensive database of protein domain movements. This involved grouping protein conformations into families, clustering the members within each family to remove redundant structures, using a Gram-Schmidt procedure to select representative pairs for each family and finally running the DynDom program to analyse protein domain movements for each family.
You can browse and search the protein families. For each family you can find information about its members, clusters and representative conformational pairs, from where you can link to the results of the individual DynDom runs.
See the following paper for details:
G. Qi, R. A. Lee, S. Hayward,
"A comprehensive and non-redundant database of protein domain movements"
Bioinformatics,21(12):2832-2838, 2005
ABSTRACT
A comprehensive and non-redundant database of protein domain movements
Guoying Qi, Richard Lee, and Steven Hayward
Motivation: The current DynDom database of protein domain motions is a user-created database that suffers from selectivity and redundancy. The aim of the analysis presented here was to overcome both these limitations and to produce both a comprehensive and non-redundant description of domain movements from structures stored in the current protein data bank.
Results: A multi-step procedure is applied that starts with grouping proteins in the structural databank into families based on sequence similarity. Multiple sequence alignment, conformational clustering, and a dimensional clustering method based on the Gram-Schmidt algorithm, are applied to members of each family to remove dynamic redundancy in their domain movements. Representative domain movements are described in terms of domains, hinge axes, and hinge-bending residues using the DynDom program. The results show that within an average family of 11.5 members there are on average only 1.31 different domain movements indicating a high redundancy in the movements these structures represent. This verifies earlier findings that domain movements are usually highly controlled. Despite the removal of this considerable redundancy the process has resulted in double the number of domain movements stored in the user-created database. The data is organised in a relational database with a web-interface.
Availability: The database can be browsed and searched at: http://www.cmp.uea.ac.uk/dyndom.
Full article in Bioinformatics